Parameterization of Polarizable Gaussian Multipole (pGM) models with the help of PyRESP_GEN
1. Introduction
Accurate description of the electrostatic interactions plays significant role in the molecular simulations. For well characterizing the electrostatic interactions, various polarizable methods have been proposed,1 such as fluctuating charge (FQ), inducible dipole (ID), and Drude oscillator and so on. Among these methods, polarization catastrophe was observed. An alternative was polarizable Gaussian Multipole (pGM) Model which utilizing the Gaussian functions with the point charges and dipoles replaced by s-orbital functions and p-orbital functions. This method could bypass the catastrophe automatically given the Gaussian functions are sufficiently diffuse. Recently, the robustness of pGM model has been validated on the interaction energies,2 many-body interaction energies together with the non-additive and additive contribution,2 and polarizability anisotropy.3 However, the input file preparation for the pGM model parameter determination is error-prone and laborious. As a consequence, we developed a program, PyRESP_GEN , which is easy-to-use and flexible.4 This program only requires ESP data file generated by espgen and could dumps 1st stage and 2nd stage file for pGM model parameterization, automatically.
In this tutorial, we will give a detailed instruction on how to make use of PyRESP_GEN4 for generating input files for pGM-ind and pGM-perm model. Then, we will show examples on the parameterization derivation using PyRESP2.
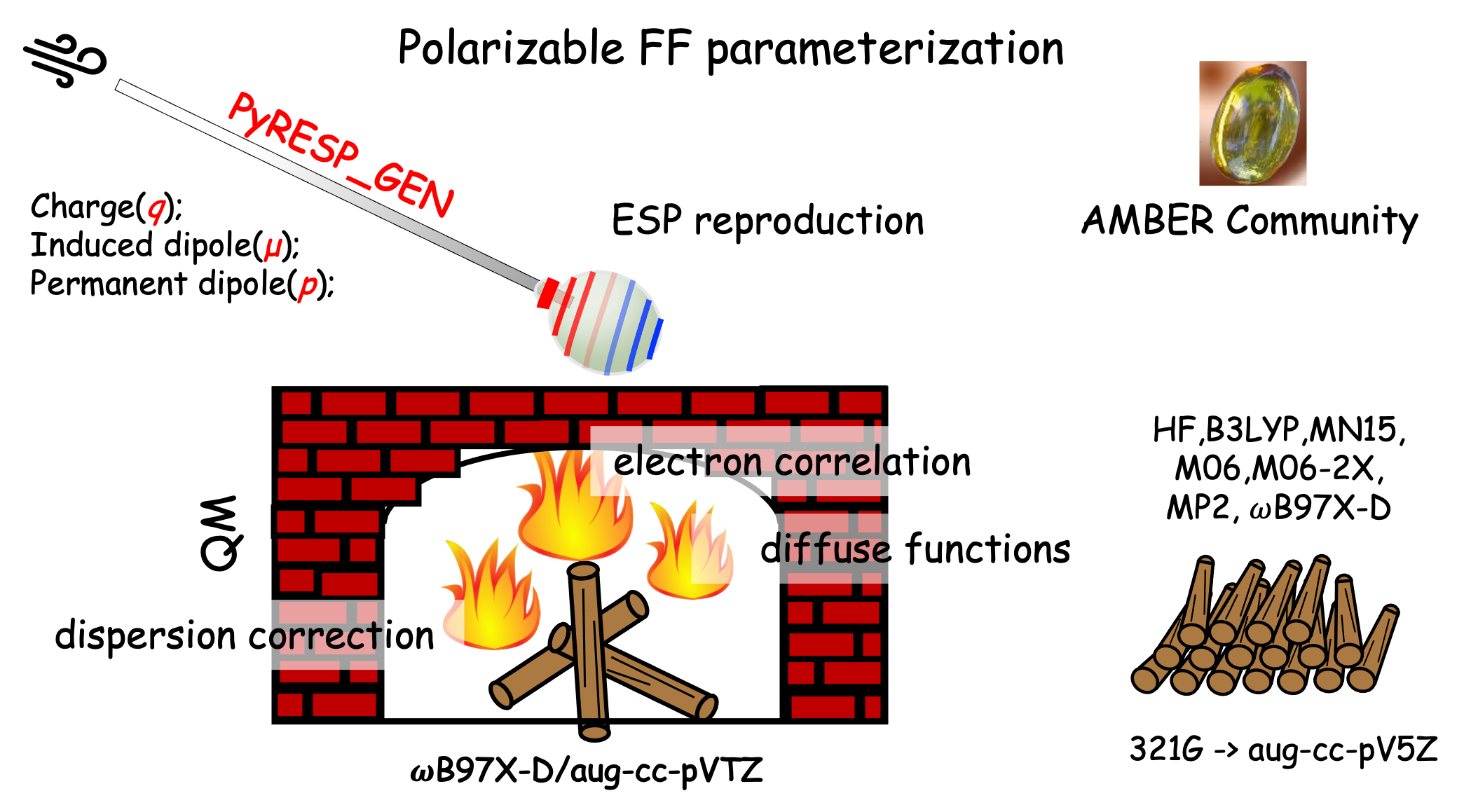
This tutorial is organized in the following order:
- Program Installation
- PyRESP_GEN
- PyRESP
- Arguments explanation of PyRESP_GEN
- Examples
- CH3SCH3
- NH4+ (charged molecule)
- CH3NH2
- References
2. Installation
2.1 Installation of PyRESP_GEN
- From source
$ git clone git@github.com:csu1505110121/pyresp_gen.git
$ cd pyresp_gen
$ conda env create -n ENV_NAME -f pyresp_gen_env.yml
$ conda activate ENV_NAME
$ export PATH=/path/to/pyresp_gen:$PATH
Setting the environment path:
- For `Zsh` Shell: Editing the ~/.zshrc and adding the following line:
export PATH=/your/path/to/pyresp_gen:$PATH
- For `Bash` User: Editing the ~/.bashrc file and adding the same line:
export PATH=/your/path/to/pyresp_gen:$PATH
$ source ~/.zshrc or source ~/.bashrc (effective immediately)
The whole package contains the following files and arranged in such hierarchical structure
$ tree
.
├── README.md
├── __init__.py
├── __pycache__
│ ├── constant.cpython-38.pyc
│ ├── constant_radii.cpython-38.pyc
│ ├── prepin.cpython-38.pyc
│ └── zmatrix.cpython-38.pyc
├── constant.py
├── constant_radii.py
├── example
│ ├── C2H6_b3lyp_321g_esp.dat
│ ├── C3H8_mp2_a4z_esp.dat
│ ├── C6H6_b3lyp_321g_esp.dat
│ ├── CH3F_ccsd_a4z_esp.dat
│ ├── CH3NH2_mp2_a4z_esp.dat
│ ├── CH3S2CH3_b3lyp_321g_esp.dat
│ ├── CH3SO2CH3_mp2_a4z_esp.dat
│ ├── CHNHOH_mp2_a4z_esp.dat
│ ├── H2_ccsd_a5z_esp.dat
│ ├── esp.dat
│ └── qiang_test_case_esp_b321.dat
├── prepin.py
├── pyresp_gen.py
├── pyresp_gen_env.yml
└── zmatrix.py
The necessary files are constant.py,constant_radii.py,prepin.py,pyresp_gen.py,and zmatrix.py.
Core functions are all stored in the file zmatrix.py. Some test examples could be found in directory example.
2.2 Installation of PyRESP
- From source
$ git clone git@github.com:ShijiZ/PyRESP.git $ export PATH=/path/to/PyRESP:$PATH Setting the environment path: - For `Zsh` User: Editing the ~/.zshrc and adding the following line: export PATH=/your/path/to/PyRESP:$PATH - For `Bash` User: Editing the ~/.bashrc file and adding the same line: export PATH=/your/path/to/PyRESP:$PATH $ export HOME_PYRESP=/your/path/to/PyRESP/polarizability $ source ~/.zshrc or source ~/.bashrc (effective immediately)
The whole package contains the following files
$ tree
.
├── polarizability
│ └── pGM-pol-2016-09-01
├── py_resp.py
py_resp.py is the main script, and pGM-pol-2016-09-01 stores the polarizability parameters derived by Wang et al.3
3. Arguments explanation
3.1 PyRESP_GEN
Usage: pyresp_gen.py [-h] --espdat ESPDAT [--Istage ISTAGE] [--IIstage IISTAGE] [--ptype PTYPE] [--dtype DTYPE] [--nmol NMOL]
[--charge CHARGE] [--QWT1 QWT1] [--QWT2 QWT2] [--PWT1 PWT1] [--PWT2 PWT2] [--EXC12 EXC12] [--EXC13 EXC13]
[--DEPTH DEPTH] [--verbose VERBOSE] [--strategy STRATEGY]
Necessary arguments:
--espdat or -i ESPDAT: Input file of esp data
Optional arguments:
-
-hor--help: Show help message and exit -
--Istageor-f1ISTAGE: Output filename of 1st stage (default:pyrespgen.1st) -
--IIstageor-f2IISTAGE: Output filename of 2nd stage (default:pyrespgen.2nd) -
--ptypeor-pPTYPE: Polarization type: xchg| xind|xperm| xperm-v(default:chg) -
--dtypeor-dDTYPE: Damping function type: xadditive| xapplequist| xtinker| xexp| xlinear(default:additive) -
--nmolor-nNMOL: Number of conformations (default:1) -
--chargeor-qCHARGE: Total charge for this structure or conformer (default:0) -
--QWT1or-qwt1QWT1: charge constraint ($a_q$) for 1st stage; (default:0.0005) -
--QWT2or-qwt2QWT2: charge constraint ($a_q$) for 2nd stage; (default:0.001) -
--PWT1or-pwt1PWT1: permanent dipoles ($a_p$) for 1st stage; (default:0.0001) -
--PWT2or-pwt2PWT2: permanent dipoles ($a_p$) for 2nd stage; (default:0.0005) -
--EXC12or-exc12EXC12: include (0) or exclude (1) 1-2 interaction (default:0) -
--EXC13or-exc13EXC13: include (0) or exclude (1) 1-3 interaction (default:0) -
--DEPTHor-depthDEPTH: Maximum depth for searching equivalent atoms (default:3) -
--verboseor-vVERBOSE: Print verbose information (default:0) -
--strategyor-strategySTRATEGY: Strategy for pGM-perm, only for debugging (default:2)
3.2 PyRESP
Usage: py_resp.py [-O] -i input -o output [-q qin] [-ip polariz] -t qout -e espot -s esout
Optional arguments:
-
-h,--help: Show this help message and exit -
-O: Overwrite output files if they exist -i,--inputINPUT type: input, required; description: input of general information-
-e,--espotESPOT type: input, required; description: input of ESP and coordinates -
-q,--qinQIN type: input, optional; description: replacement parameters -
-o,--outputOUTPUT type: output, always produced; description: output of results -
-t,--qoutQOUT type: output, always produced; description: output of parameters -
-s,--esoutESOUT type: output, optional; description: generated ESP values for new parameters -ipPOLARIZ, –polariz POLARIZ type: input, optional; description: atomic polarizabilities
4. Examples
4.1 Case 1: CH3SCH3
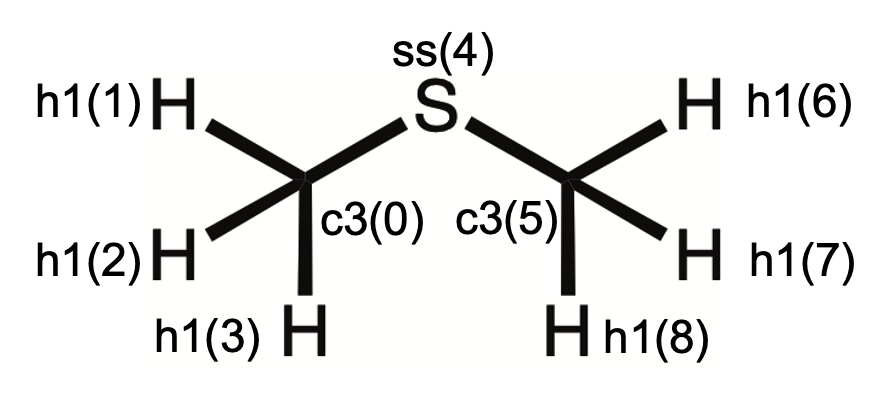
4.1.1 Generation of electrostatic potentials with Gaussian
Step 1: input file generation
(filename: CH3SCH3.com)
%chk=CH3SCH3_a2z (specify the chk file name)
%nproc=28 (specify the num. of process )
%mem=28GB (specify the memory)
#b3lyp/aug-cc-pvdz (methods/basis sets)
#SCF=tight
#maxdisk=200GB
#Pop=MK iop(6/33=2) iop(6/42=6) iop(6/43=20)
#IOP(5/87=12)
#density=current
C3 MP2/6-311++G(d,p) coordinates (optional)
0 (charge) 1. (multiplicity)
C (elem) -1.361796871769 (x coor) -0.515789669798 (y coor) -0.000057835254 (z coor)
H (elem) -2.293815474604 (x coor) 0.053063245903 (y coor) -0.000100910602 (z coor)
H (elem) -1.338738381082 (x coor) -1.144434093987 (y coor) -0.893950138079 (z coor)
H (elem) -1.338815515689 (x coor) -1.144425446053 (y coor) 0.893842578094 (z coor)
S (elem) 0.000000000000 (x coor) 0.666316788502 (y coor) -0.000005794613 (z coor)
C (elem) 1.361796871989 (x coor) -0.515789668820 (y coor) 0.000066888412 (z coor)
H (elem) 2.293815474803 (x coor) 0.053063247384 (y coor) 0.000108971075 (z coor)
H (elem) 1.338733743617 (x coor) -1.144425283672 (y coor) 0.893965305093 (z coor)
H (elem) 1.338820151777 (x coor) -1.144434253910 (y coor) -0.893827410725 (z coor)
(blank is needed here)
(blank is needed here)
Input file: CH3SCH3.com
Step 2: Execute Gaussian
# Make sure you have install Gaussian 16 or 09 correctly.
$ g16 CH3SCH3.com &
Output file: CH3SCH3.log
Step 3: Convert the log file into ESP file
$ espgen_junmei -i CH3SCH3.gout -o CH3SCH3.dat -p 1
## You will see following files:
## ESPGEN.TMP and CH3SCH3.dat
Esp data file: CH3SCH3.dat
The dat file head is shown as below:
1 9 (num. of atoms) 3780 (num. of grids) 0 MOL 9 3780
2 -2.5734232E+00 (x coor) -9.7470177E-01 (y coor) -1.0960411E-04 (z coor) 6 (elem) c3 (atom type) C1
3 -4.3346818E+00 (x coor) 1.0027453E-01 (y coor) -1.9086232E-04 (z coor) 1 (elem) h1 (atom type) H1
4 -2.5298480E+00 (x coor) -2.1626667E+00 (y coor) -1.6893205E+00 (z coor) 1 (elem) h1 (atom type) H2
5 -2.5299954E+00 (x coor) -2.1626497E+00 (y coor) 1.6891183E+00 (z coor) 1 (elem) h1 (atom type) H3
6 0.0000000E+00 (x coor) 1.2591566E+00 (y coor) -1.1338356E-05 (z coor) 16 (elem) ss (atom type) S1
7 2.5734232E+00 (x coor) -9.7470177E-01 (y coor) 1.2661164E-04 6 c3 C2
8 4.3346818E+00 (x coor) 1.0027453E-01 (y coor) 2.0598013E-04 (z coor) 1 (elem) h1 (atom type) H4
9 2.5298404E+00 -2.1626497E+00 1.6893489E+00 1 h1 H5
10 2.5300029E+00 -2.1626667E+00 -1.6890881E+00 1 h1 H6
11 -1.2030000E-03 (esp value) -3.3327472E+00 (esp x coor) 3.4048894E-01 (esp y coor) 3.6662366E+00 (esp z coor)
4.1.2 PyRESP_GEN generation
Step 1: Input file generation
# generating input file for RESP
$ cd 3-resp
$ pyresp_gen.py -i CH3SCH3.dat -p chg
# cd 4-ind
$ pyresp_gen.py -i CH3SCH3.dat -p ind
# cd 5-perm
$ pyresp_gen.py -i CH3SCH3.dat -p perm
4.2 Case 2: NH4+
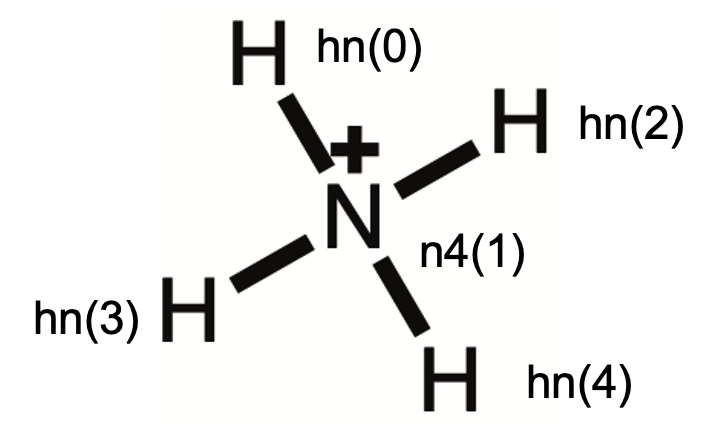
4.2.1 Generation of electrostatic potentials with Gaussian
Step 1: Input file generation
%chk=NH4+_b3lyp_a2z_esp
%nproc=28
%mem=28GB
#b3lyp/aug-cc-pvdz
#SCF=tight
#maxdisk=200GB
#Pop=MK iop(6/33=2) iop(6/42=6) iop(6/43=20)
#IOP(5/87=12)
#density=current
Coordinates from nh4_m6311_opt.chk
1 1
H -0.951190136213 0.348537851523 0.152959323805
N -0.000052158259 0.000090031932 0.000048013550
H 0.142159883375 -0.855519120874 0.545365091572
H 0.133112562719 -0.203821543598 -0.995003830076
H 0.676282797933 0.710172589425 0.296343319850
Step 2: Execute Gaussian
$ g16 NH4+.com &
Step 3: Convert the log file into ESP file
$ espgen_junmei -i NH4+.gout -o NH4+.dat -p 1
## You will see following files:
## ESPGEN.TMP and NH4+.dat
4.2.2 PyRESP_GEN generation
Step 1: Input file generation
*** Caution ***
*** for charged species ***
*** you need arg `-q` to specify the charge manually ***
# generating input file for RESP
$ cd 3-resp
$ pyresp_gen.py -i CH3SCH3.dat -p chg -q 1
# cd 4-ind
$ pyresp_gen.py -i CH3SCH3.dat -p ind -q 1
# cd 5-perm
$ pyresp_gen.py -i CH3SCH3.dat -p perm -q 1
4.3 Case 3: CH3NH2
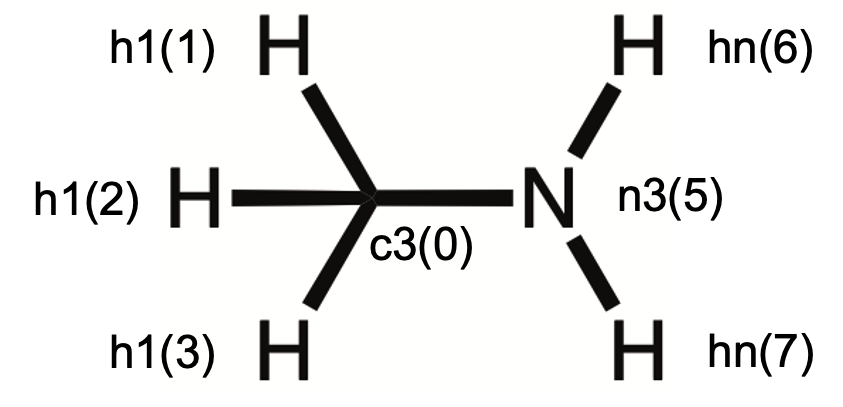
4.3.1 Generation of electrostatic potentials with Gaussian
Step 1: Input file generation
%chk=CH3NH2_b3lyp_a2z_esp
%nproc=28
%mem=28GB
#b3lyp/aug-cc-pvdz
#SCF=tight
#maxdisk=200GB
#Pop=MK iop(6/33=2) iop(6/42=6) iop(6/43=20)
#IOP(5/87=12)
#density=current
B2 MP2/6-311++G(d,p) coordinates
0 1
C -0.708095503398 -0.000000893539 0.017712823484
H -1.112963870065 -0.880513863166 -0.486271271246
H -1.074379816824 -0.000087389127 1.053630363354
H -1.112943454187 0.880611141673 -0.486114099690
N 0.750287956697 -0.000000780330 -0.122136797163
H 1.148419546341 0.813654667261 0.333717877871
H 1.148424918244 -0.813653733099 0.333717768949
Step 2: Execute Gaussian
$ g16 CH3NH2.com &
Step 3: Convert the log file into ESP file
$ espgen_junmei -i CH3NH2.gout -o CH3NH2.dat -p 1
## You will see following files:
## ESPGEN.TMP and CH3NH2.dat
4.3.2 PyRESP_GEN generation
Step 1: Input file generation
# generating input file for RESP
$ cd 3-resp
$ pyresp_gen.py -i CH3NH2.dat -p chg -q 0 -v 1
# cd 4-ind
$ pyresp_gen.py -i CH3NH2.dat -p ind -q 0 -v 1
# cd 5-perm
$ pyresp_gen.py -i CH3NH2.dat -p perm -q 0 -v 1
Since argment -v is activated, you will see verbose output information on the screen as below
Distance Map in the unit of angstrom (A)
c3 h1 h1 h1 n3 hn hn
6 0.000 1.092 1.099 1.092 1.465 2.051 2.051
1 1.092 0.000 1.774 1.761 2.093 2.942 2.406
1 1.099 1.774 0.000 1.774 2.171 2.474 2.474
1 1.092 1.761 1.774 0.000 2.093 2.406 2.942
7 1.465 2.093 2.171 2.093 0.000 1.014 1.014
1 2.051 2.942 2.474 2.406 1.014 0.000 1.627
1 2.051 2.406 2.474 2.942 1.014 1.627 0.000
Equivalent Atom Map:
+ 1 denotes equil
+ 0 denotes not equil
6 1 1 1 7 1 1
6 1.0 0.0 0.0 0.0 0.0 0.0 0.0
1 0.0 1.0 1.0 1.0 0.0 0.0 0.0
1 0.0 1.0 1.0 1.0 0.0 0.0 0.0
1 0.0 1.0 1.0 1.0 0.0 0.0 0.0
7 0.0 0.0 0.0 0.0 1.0 0.0 0.0
1 0.0 0.0 0.0 0.0 0.0 1.0 1.0
1 0.0 0.0 0.0 0.0 0.0 1.0 1.0
#------------------------------------------------------------#
Detailed Info. derived from the *Distance Map*:
Idx. Center Idx. Bonded (Atm. Type Bonded)
0 1 ( h1) 2 ( h1) 3 ( h1) 4 ( n3)
1 0 ( c3)
2 0 ( c3)
3 0 ( c3)
4 0 ( c3) 5 ( hn) 6 ( hn)
5 4 ( n3)
6 4 ( n3)
Detailed Info. for -CH2-:
NO -CH2- FRAGMENT WAS DETECTED *PLEASE CHECK STRUCTURE*
Detailed Info. for -CH3:
Index for Carbon was listed below:
o 0
Equivalent HEAVY atom index:
NO EQUIVALENT HEAVY ATOM WAS DETECTED!
Equivalent HYDROGEN atom index for -CH2-:
NO -CH2- FRAGMENT WAS DETECTED!
Equivalent HYDROGEN atom index for -CH3-:
EQUIVALENT HYDROGEN INDEX (-CH3):
Idx. Idx.
1 2
1 3
Equivalent HYDROGEN atom index exclude -CH2- and -CH3
EQUILVALENT HYDROGEN INDEX (EXCLUDING -CH2-, -CH3)
Idx. Idx.
5 6
#------------------------------END------------------------------#
The first matrix is distance map in the unit of angstrom, it defines the distance formed between any two paired atoms;
The second matrix is equivalent atom map which defines the equivalence between two atom, 1 for equivalent and 0 for inequivalent;
Following message is detailed information derived from Distance Map ;
Taking molecule CH3NH2 as an example:
The first atom with index 0 is bonded with four atoms, namely, 1(h1), 2(h1), 3(h1), and 4(n3), the str in the parentheses is the atom type derived from espgen;
Then, message shows the information of function group -CH2- and -CH3.
In this molecule, only one -CH3 group was found, and index of atom Carbon is 0;
Additional information is the equivalence information for heavy atoms (excluding -CH2-, -CH3) , -CH2-, -CH3, and hydrogen atoms (excluding -CH2-,-CH3), -CH2-, -CH3.
For example, in CH3NH2 molecule, equivalent H atoms of -CH3 is as below:
1(h1) == 2(h1)
1(h1) == 3(h1)
Since no -CH2- was found in CH3NH2, no equivalent H atom index of -CH2- was detected.
Equivalent hydrogen index (excluding -CH2-, -CH3) was found
5(hn) == 6(hn)
4.4 PyRESP execution
4.4.1 For RESP model
# Stage one
$ py_resp.py -O\
$ -i pyrespgen.1st\ (1st file generated by PyRESP_GEN)
$ -o pyrespgen.1st.out\ (output filename specified by yourself)
$ -e ESPDATA.dat\ (esp data file generated by ESPGEN)
$ -s FITTED_ESPDATA.1st.esp\ (filename of fitted esp data)
$ -t FITTED_CHG.1st.chg\ (filename of fitted chg data)
# Stage two
$ py_resp.py -O \
$ -i pyrespgen.2nd\ (2nd file generated by PyRESP_GEN)
$ -o pyrespgen.2nd.out\
$ -e ESPDATA.dat\ (esp data file generated by ESPGEN)
$ -s FITTED_ESPDATA.2nd.esp\ (filename of fitted esp data)
$ -t FITTED_ESPDATA.2nd.chg\ (filename of fitted chg data)
$ -q FITTED_CHG.1st.chg (1st fitted chg data)
4.4.2 For pGM-ind and pGM-perm model
# Stage one
$ py_resp.py -O\
$ -i pyrespgen.1st\ (1st file generated by PyRESP_GEN)
$ -o pyrespgen.1st.out\ (output filename specified by yourself)
$ -e ESPDATA.dat\ (esp data file generated by ESPGEN)
$ -ip ${HOME_PYRESP}/pGM-pol-2016-09-01\ (polarizability parameters)
$ -s FITTED_ESPDATA.1st.esp\ (filename of fitted esp data)
$ -t FITTED_CHG.1st.chg\ (filename of fitted chg data)
# Stage two
$ py_resp.py -O \
$ -i pyrespgen.2nd\ (2nd file generated by PyRESP_GEN)
$ -o pyrespgen.2nd.out\
$ -e ESPDATA.dat\ (esp data file generated by ESPGEN)
$ -ip ${HOME_PYRESP}/pGM-pol-2016-09-01\ (polarizability parameters)
$ -s FITTED_ESPDATA.2nd.esp\ (filename of fitted esp data)
$ -t FITTED_ESPDATA.2nd.chg\ (filename of fitted chg data)
$ -q FITTED_CHG.1st.chg (1st fitted chg data)
References
[1] Qiang Zhu, Yang Ge, Wei Li,* and Jing Ma*. Treating Polarization Effects in Charged and Polar Bio-Molecules Through Variable Electrostatic Parameters. J. Chem. Theory Comput. 2023, 19, 2, 396–411;
[2] Shiji Zhao, Haixin Wei, Piotr Cieplak, Yong Duan,* and Ray Luo.* PyRESP: A Program for Electrostatic Parameterizations of Additive and Induced Dipole Polarizable Force Fields. J. Chem. Theory Comput. 2022, 18, 6, 3654–3670;
[3] Junmei Wang*, Piotr Cieplak, Ray Luo, and Yong Duan*. Development of Polarizable Gaussian Model for Molecular Mechanical Calculations I: Atomic Polarizability Parameterization To Reproduce ab Initio Anisotropy. J. Chem. Theory Comput. 2019, 15, 2, 1146–1158;
[4] Qiang Zhu, Yongxian Wu, Shiji Zhao, Piotr Cieplak, Yong Duan,* Ray Luo.* Streamlining and Optimizing Strategies of Electrostatic Parameterization J. Chem. Theory Comput. 2023, 19,18, 6353-6365.